Free Energy Calculations
Methodology and applications of free energy calculations
All chemical equilibria are governed by the difference in free energy between various states. Examples are free energy differences between different conformations of a biomolecule or the free energy difference between the bound and unbound state of a ligand and its pharmaceutical target. In our group we try to develop methods to efficiently and accurately calculate free energy differences for such processes, using statistical mechanics and molecular simulation. In recent years, we have used the one step perturbation method to calculate relative binding free energies for series of compounds. This method is currently being further developed on a set of trypsin inhibitors and applied to a crop protection target.
Accelerated Enveloping Distribution Sampling (A-EDS)
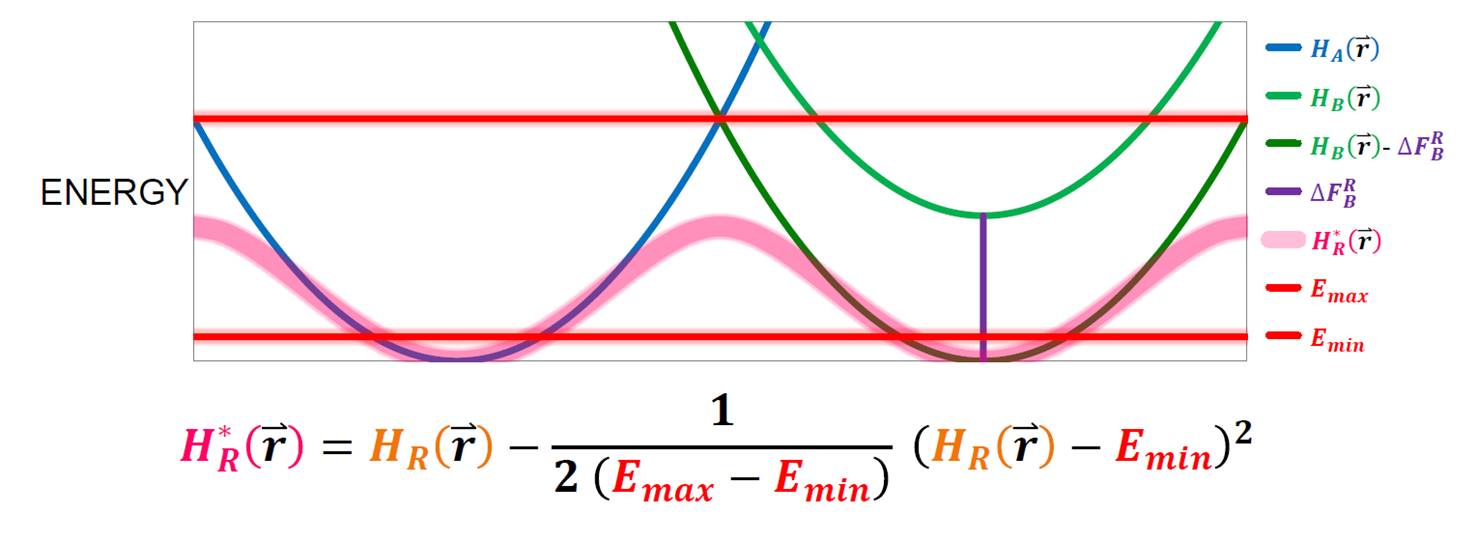
Accelerated Enveloping Distribution Sampling (A-EDS)
We are developing an extension to the original EDS Hamiltonian proposed by Christ et al. Large energy barriers between end-states are decreased by smoothening with a harmonic potential, similar to accelerated MD proposed by McCammon and coworkers. This allows for efficient sampling of multiple end-states while original energy minima are preserved. Like one-step perturbation approaches, the method is applied to diverse free-energy calculations and can be used to calculate multiple free-energy differences from a single simulation, including charge-changes.
J.W. Perthold and C. Oostenbrink
Accelerated Enveloping Distribution Sampling: Enabling Sampling of Multiple End States while Preserving Local Energy Minima
J Phys Chem B. 122 (2018) 5030-5037
doi: 10.1021/acs.jpcb.8b02725
One-step perturbation and third-power fitting
One-step perturbation and third-power fitting
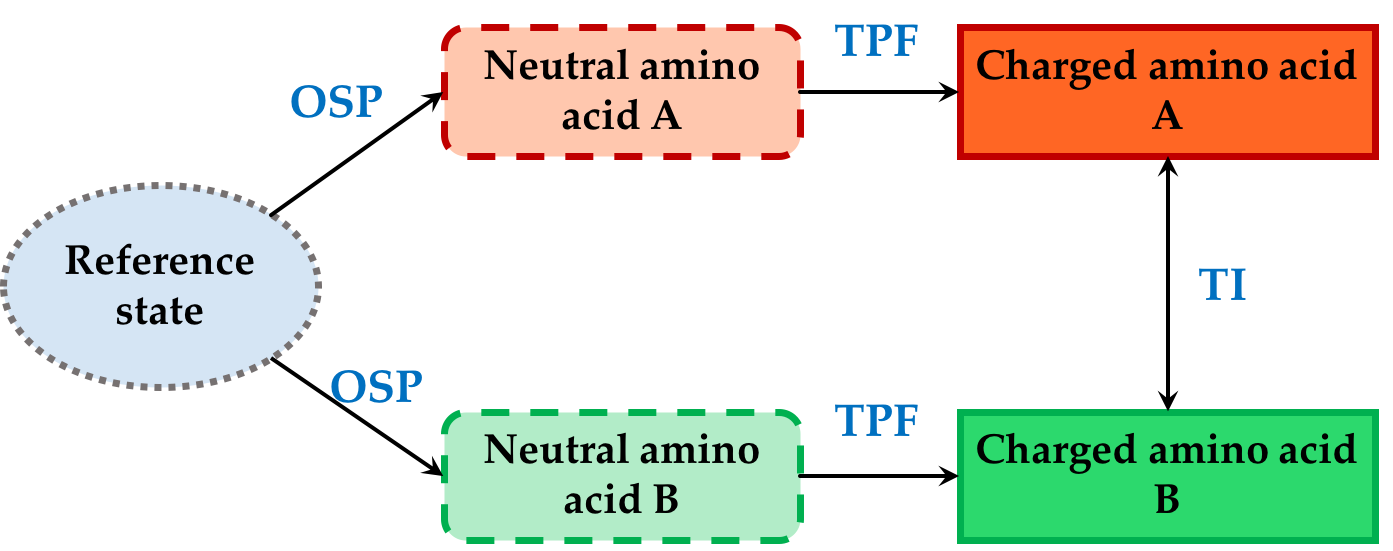
Mutation of one amino acid into all other possible amino acids at a certain position in the protein, also known as saturation mutagenesis, can have a great impact on protein stability, binding affinity, protein function or its expression per se. Our aim is to accurately and efficiently predict the free energy of mutation of amino acids in proteins, i.e. their consequential effects on above mentioned properties. We are using a combination of One Step Perturbation (OSP) to calculate the free energy of changes in Van der Waals contributions and Third Power Fitting (TPF) for calculation of Coulombic contributions to the free energy.
Protein-ligand interaction

Protein-ligand interaction
Methods like local elevation and umbrella sampling are are used to compute the actual dissociation and association paths of the ligands binding to proteins. One challenge in path-sampling methods is to achieve reversible binding because it is easy for the ligand to get stuck at the wrong side of the protein in the association process driven by conventional distance restraints. We devised distance field distance restraints as an alternative reaction coordinate. By mapping distances on a grid and penalizing
pathways passing through the protein we considerably improve reversible binding.
A. de Ruiter and C. Oostenbrink
Protein-Ligand Binding from Distancefield Distances and Hamiltonian Replica Exchange Simulations
J. Chem. Theory Comput. 9 (2012) 883-892
doi: 10.1021/ct300967a
Protein-protein interactions
Protein-protein interactions
The calculation of protein-protein binding free energies is a difficult endeavor given the complicated nature of interactions and possible conformational changes associated with binding reactions. We develop methods using perturbed distance restraints in combination with enhanced sampling techniques to accurately calculate binding free energies of proteins and other complex biomolecules. Particular attention is paid to the demonstration of reversibility of the simulated reactions and to convergence of the calculated free-energy values.
J.W. Perthold and C. Oostenbrink
Simulation of Reversible Protein-Protein Binding and Calculation of Binding Free Energies Using Perturbed Distance Restraints
J Chem Theory Comput. 13 (2017) 5697-5708
doi: 10.1021/acs.jctc.7b00706

Charging free-energy calculations

Charging free-energy calculations
The calculation of charging-free energies from atomistic classical molecular dynamics simulation is a difficult task, because the results are dependent on the chosen spatial boundary conditions and effective electrostatic interaction scheme. The associated artifacts in the raw simulation results are well understood if the solute to be charged consists of a single energy group. However, conceptual ambiguities in the calculation of charging free energies (as well as virial and energy) arise if the solute consists of multiple energy groups. Therefore, a reformulation of electrostatic interactions in the GROMOS program for (bio-)molecular simulation is undertaken. Through the absence of a self term, the calculation of charging free energies (as well as virial and energy) in systems presenting a net charge can thus be performed in an unambiguous fashion. The corresponding new electrostatic interaction functions and partitioning into multiple energy groups is implemented in the GROMOS program for both lattice-sum and cutoff-truncated reaction-field electrostatics.