Force-field development
Force-field development
The careful optimisation of force field parameters is essential for any classical simulation. Nonbonded interactions can currently be parameterised against free energies of solvation in various media. For ionic groups, there are several conceptual difficulties that need to be overcome, in which great progress has been made recently.
Atomistic force-field development and testing
Atomistic force-field development and testing
The GROMOS force field 54A8 is the first of its kind to contain nonbonded parameters for charged amino acid side chains that were derived in a rigorously thermodynamic fashion, namely a calibration against hydration free energies in the infinitely dilute regime. The raw hydration free energies calculated from atomistic simulations are affected by electrostatic and finite-size artifacts, and corrections are applied to reach methodological independence prior to comparison with the experimental values.
M. Reif, P. Hünenberger and C. Oostenbrink
New interaction parameters for charged amino acid side chains in the GROMOS force field
J. Chem. Theory and Comp. (2012) 8, 3705 - 3723
doi: 10.1021/ct300156h

Optimization strategies
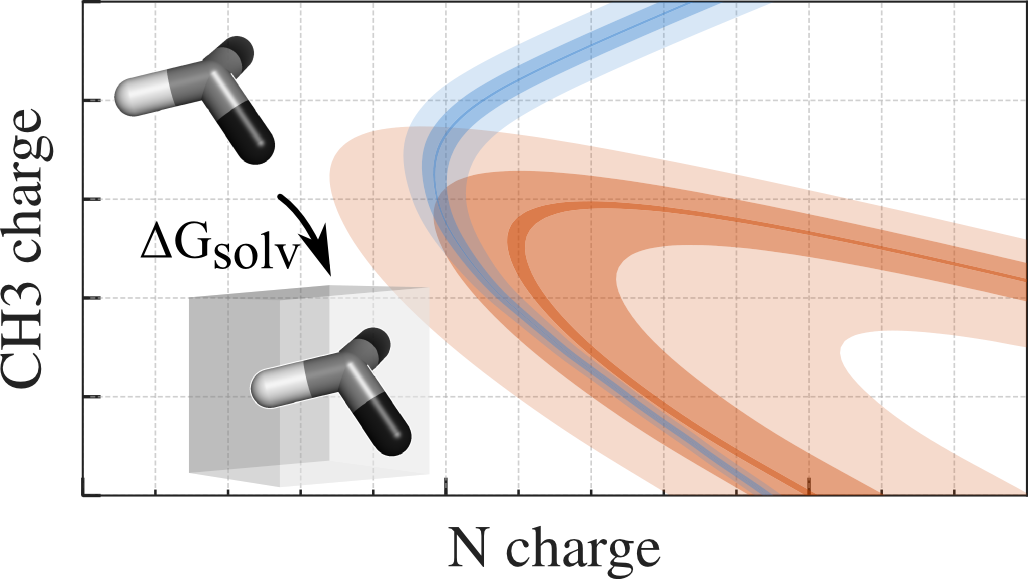
Optimization strategies
The optimization of force-field parameters is a multi-dimensional problem. The simulation effort can be significantly reduced by a combination of one-step perturbation (OSP) and steepest descent minimization of free-energies in parameter space. We have applied this to optimize the charges of small amines in water, chloroform and tetrachloromethane, but it can well be extended to other parameters like the Lennard-Jones C6 and C12 terms.
M. Pechlaner, M. Reif and C. Oostenbrink
Reparametrisation of united-atom amine solvation in the GROMOS force field
Mol. Physics (2017) doi:10.1080/00268976.2016.1255797
Parameterization against NMR observables

The force field defines the way atoms and molecules interact with each other in molecular dynamics simulations. Observables from NMR experiments (J-coupling values, NOE distances,...) give a very detailed picture of the structural properties of proteins and allow us to test the molecular simulations directly against experimental data. The observations from a large number of simulations and the parameterization of force-field parameters against NMR observables for smaller molecules may guide further improvements on the force fields.