Prediction and mapping of heterosis effects using genome-wide SNP arrays in advanced inter-cross populations
SUPERVISOR: Johann SÖLKNER
PROJECT ASSIGNED TO: Tesfay GEBRESELAMA TEWELDEMEDHN
Systematic crossbreeding in plant and animal breeding is used to combine the positive attributes of parental lines as well as taking advantage of hybrid vigor or “heterosis”. Heterosis is the deviation of crossbred progeny from the average of their purebred parental lines for a trait. Heterosis is due to non-additive genetic effects, with dominance and epistatic components. The genome of a crossbred individual is a mosaic of ancestral haplotypes formed by recombination along generations. Ancestry origin at chromosome levels in admixed individuals, i.e. “local genetic ancestry”, can be used to define the components of heterosis. Recent advance in high throughput genotyping and sequencing has provided the opportunity of accurately estimating local ancestry proportions.
Prediction of heterosis effects is a major issue in agricultural research. The availability of whole-genome sequencing arrays has provided more precise information compared to pedigree relationships, encouraging the researchers to include non-additive genetic effects (dominance and epistasis) for the analysis of complex traits in genomic prediction of genetic merit of individual animals.
While a large proportion of animal products currently consumed around the world is based on hybrid or crossbred individuals, the genetic effects causing heterosis are not well elucidated. In this project, we will compare models based on breed ancestry with models based on actual genotypes of each of very many genetic markers across the genome. We focus on advanced crosses because it is not possible to accurately map effects contributing to heterosis in first or second generation crosses. We will take advantage of phenotypes and high throughput genotypes already collected in experimental populations of chicken and mouse.
Breed composition estimated at local level can be used to define heterosis components and to estimate and map genomic heterosis effects in crossbred populations. The genomic basis of heterosis effects will be elucidated using genome-wide molecular markers, by developing appropriate genetic models to predict and map effects of dominance and epistasis in advanced intercross lines of experimental chicken and mouse.
The research hypothesis states that, with high throughput genetic markers, breed composition estimated at local level (locus-specific genetic ancestry) can be used to define heterosis components and to estimate and map genomic heterosis effects in advanced crossbred populations. This project aims at dissecting the genomic basis of heterosis effects by developing appropriate genetic models to predict and map heterosis components (dominance and epistatic interaction components of heterosis). The heterosis components will be estimated and mapped in advanced intercross lines of Fayoumi × Broiler chicken provided by Iowa State University and Berlin Fat Mouse (BMI) × C57Bl/6CrlN (B6N) provided by Humboldt Universität Berlin. The animal breeding and genetics groups of these two universities are collaborators of this project.
Specifically, the project focuses on the following objectives.
A. To estimate genome wide components of heterosis, including dominance and epistatic loss, using genome-wide local genetic ancestry for economically important traits in experimental advanced inter-cross lines (AILs) of chicken and mouse.
B. To perform regional association mapping of heterosis components by defining the components based on either straight genotype information or local genetic ancestries.
C. To simulate genotypes and their effects (additive, dominance and epistasis) as well as phenotypes to evaluate competing methods for estimating and mapping heterosis effects. This can help to compare accuracy of prediction of heterosis components based on local ancestry versus genotype information.
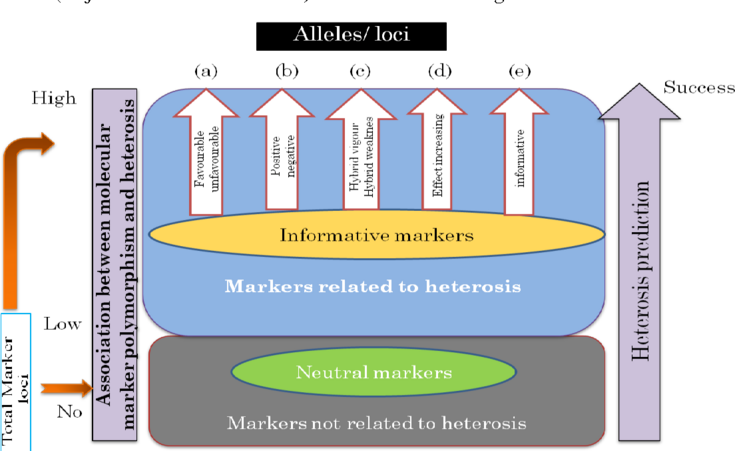
Figure 1: Concept of Heterosis Prediction Using Genomic Data
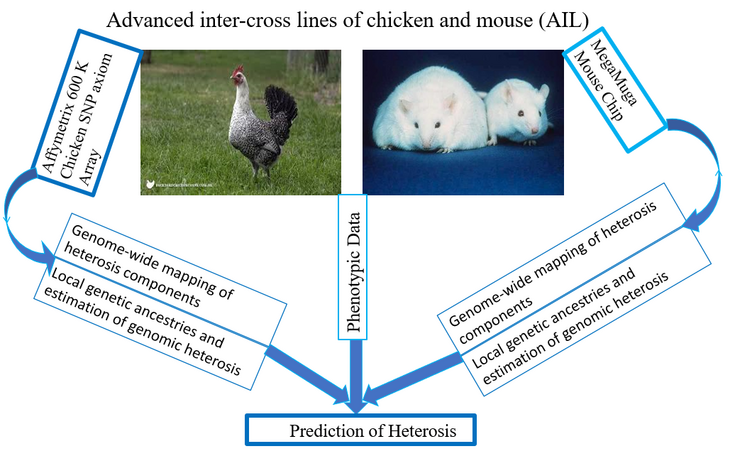
Figure 2: Schematic Diagram of the project