Structure and dynamics of biomolecules
The structure and dynamics of complex biomolecular structures are studied by molecular dynamics simulations. This gives insight into the function, stability and potential modification sites of such molecules.
Restoring affinity in a humanized antibody
Restoring affinity in a humanized antibody
Antibodies recovered from mice have a high risk of evoking an anti-mouse immune response in humans. To make them suitable for therapeutic application, murine anti-bodies have to be humanized which often leads to loss in affinity. We simulated a set of humanized Ab2/3H6 mutants (in the absence of binding partner 2F5) and from the conformational ensemble of the antigen-binding site were able to predict a minimal set of mutations which proved to restore binding affinity.
C. Margreitter, P. Mayrhofer, R. Kunert and C. Oostenbrink
Antibody humanization by molecular dynamics simulations—in-silico guided selection of critical backmutations
J. Mol. Recognit. 29 (2016) 266-275
doi: 10.1002/jmr.2527

Ion binding to chlorite dismutases
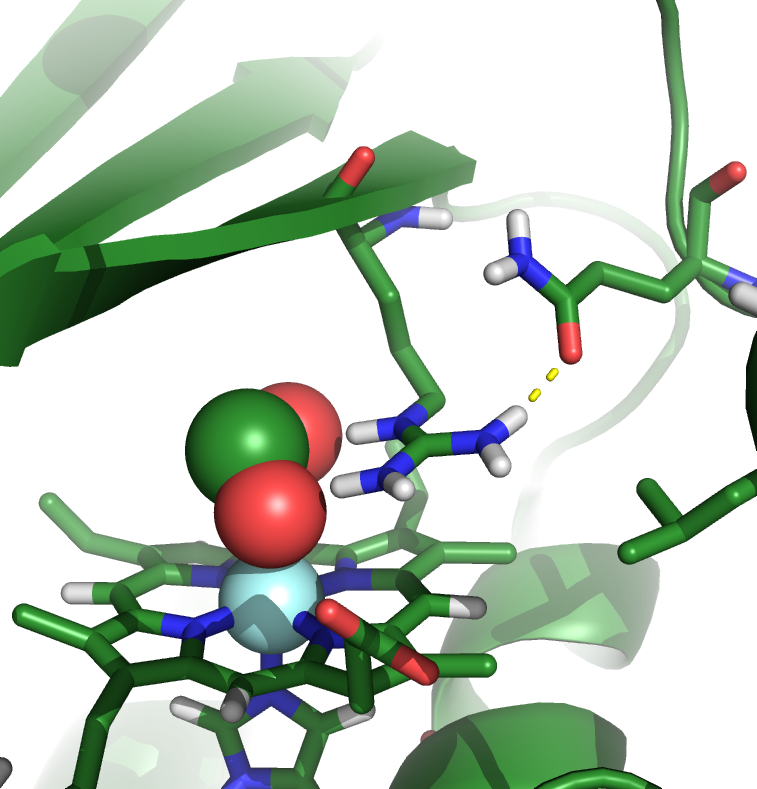
Ion binding to chlorite dismutases
Chlorite dismutases are heme-containing proteins with the remarkable ability of producing molecular oxygen from the toxic pollutant chlorite (ClO2-). The active site cavity is accessible by a narrow channel and contains only one charged residue, and arginine which seems to play a role in the guidance of the ions. The putative reaction pathway involves an intermediate hypochlorite (CLO-) which has been suggested to be responsible for the irreversible inactivation of the enzyme in in-vitro experiments. Using MD simulations and free-energy calculations we study the effect of mutations inside the active site on binding of CLO2- and CLO- to the enzyme to further our understanding of the reaction mechanism.
A. Sündermann, M.M. Reif, S. Hofbauer, C. Obinger and C. Oostenbrink
Investigation of ion binding in chlorite dismutases by means of molecular dynamics simulations
Biochemistry 53 (2014) 4869 - 4879
doi: 10.1021/bi500467h
Water-mediated biomolecular interactions
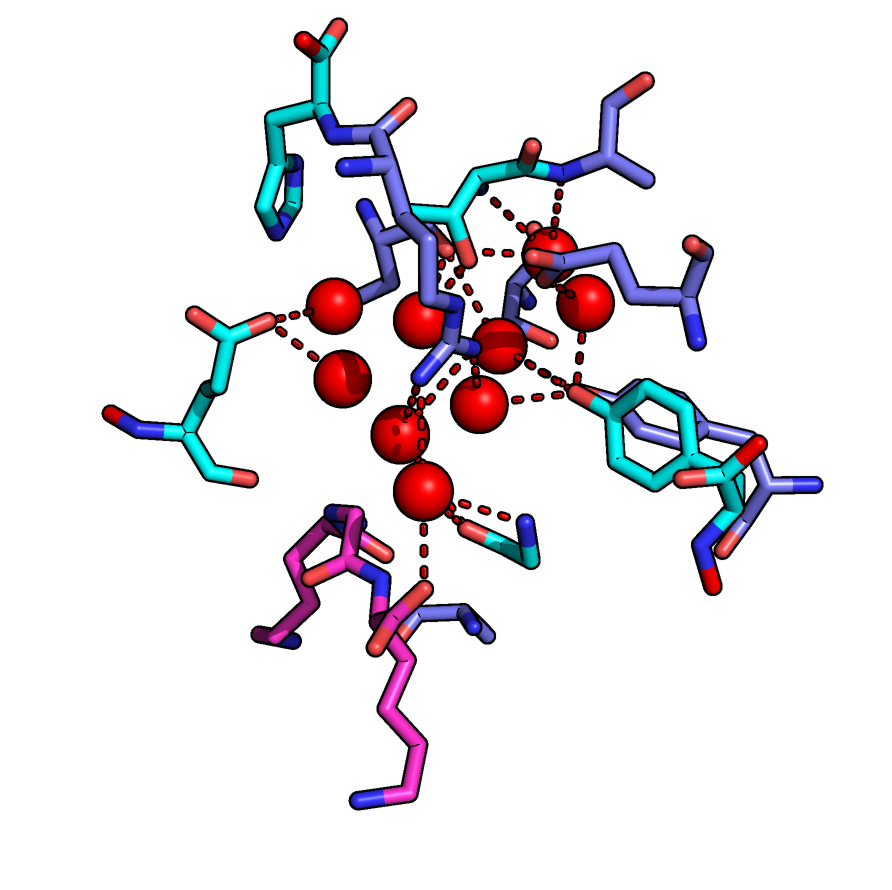
Water-mediated biomolecular interactions
The periplasmic oligopeptide binding protein A (OppA) represents a well-known example of water-mediated ligand binding. We chose ligands of OppA with a high structural similarity yet remarkably different binding affinity and use them as a test case to perform binding free energy calculations using several methods such as Thermodynamic Integration and Enveloping Distribution Sampling. We are developing and testing approaches which combine these methods in order to be able to calculate the influence of structurally relevant water molecules on binding independently, on top of ligand changes or protein mutations.
M. Maurer, S.B.A. de Beer and C. Oostenbrink
Calculation of relative binding free energy in the water-filled active site of oligopeptide-binding protein A
Molecules 21 (2016), 499
doi: 10.3390/molecules21040499
Bioremediation
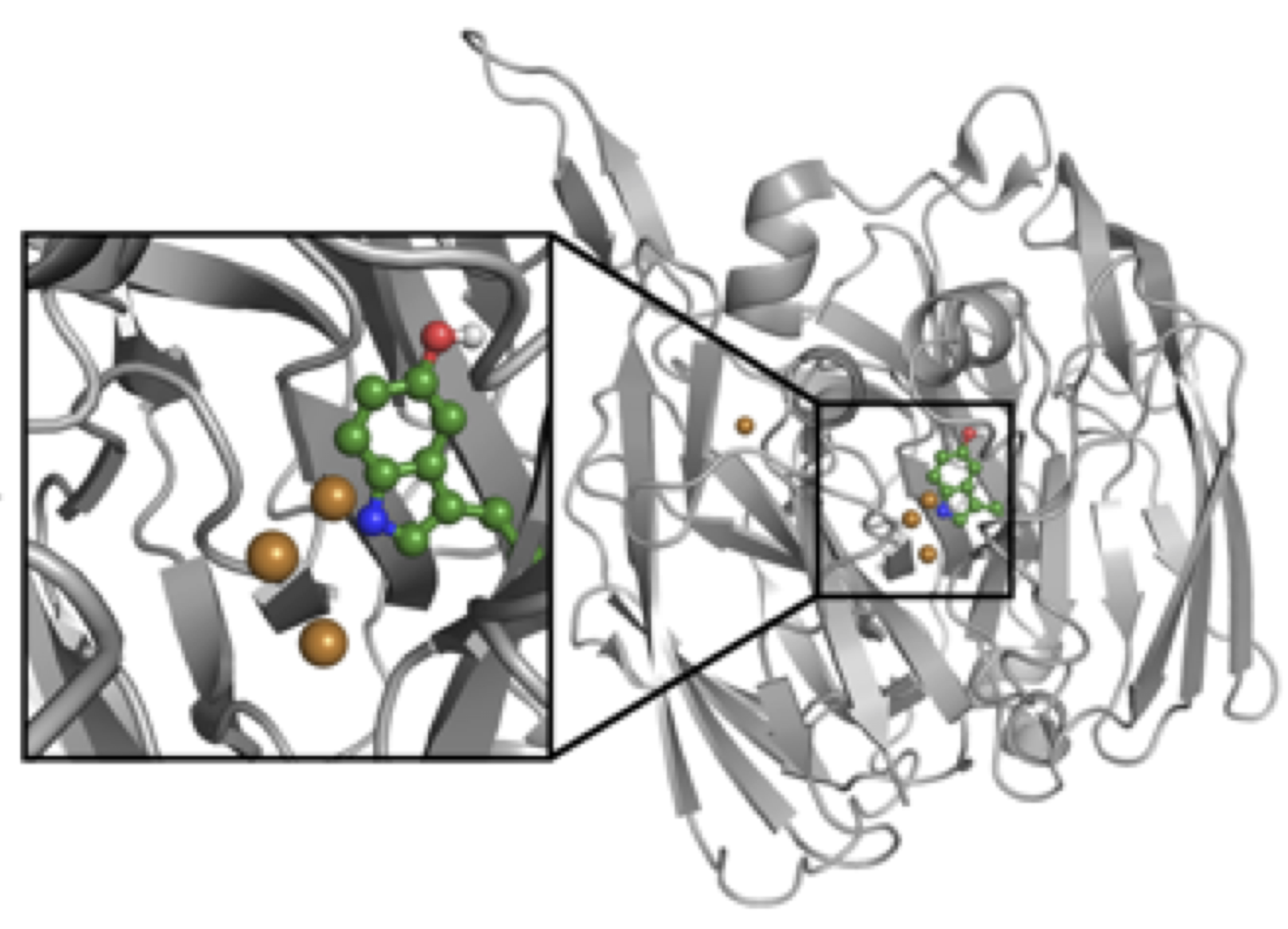
Bioremediation
Extracellular enzymes from bacteria, fungi and plants have a great potential in remediation of a broad range of pollutants. One of the greatest bottlenecks in their wider applications lies in their reduced effectiveness when utilized directly in soils at polluted sites. Using extensive MD simulations, we ask how enzyme structure and dynamics depend on: 1) different composition of soil organic matter, 2) different conditions (temperature, pH and water content) and 3) level and type of oxidative modifications.
Glycosylated proteins
Glycosylated proteins
The determination of the exact molecular structure of glycosylated proteins is often a challenge due to the inherent flexible nature of glycan units. Even if the molecular structure has been resolved, they tend to lack a complete representation of the glycan units. To describe these complex structures at a time and a space resolution that is not attainable with experiments, we generate relevant structures and simulate them to gain insight about the importance of glycan units on function. For the generation of relevant glycan units, we use an enhanced sampling method, local elevation with umbrella sampling (LEUS) to search the relevant conformational space. After having reliable conformations of them, we build glycosylated proteins by modeling glycan structures onto available deglycosylated protein structures with different techniques in the absence of any experimental data. To gain insight about their functionality, we simulate glycosylated proteins either with different glycoforms or in the absence of them.
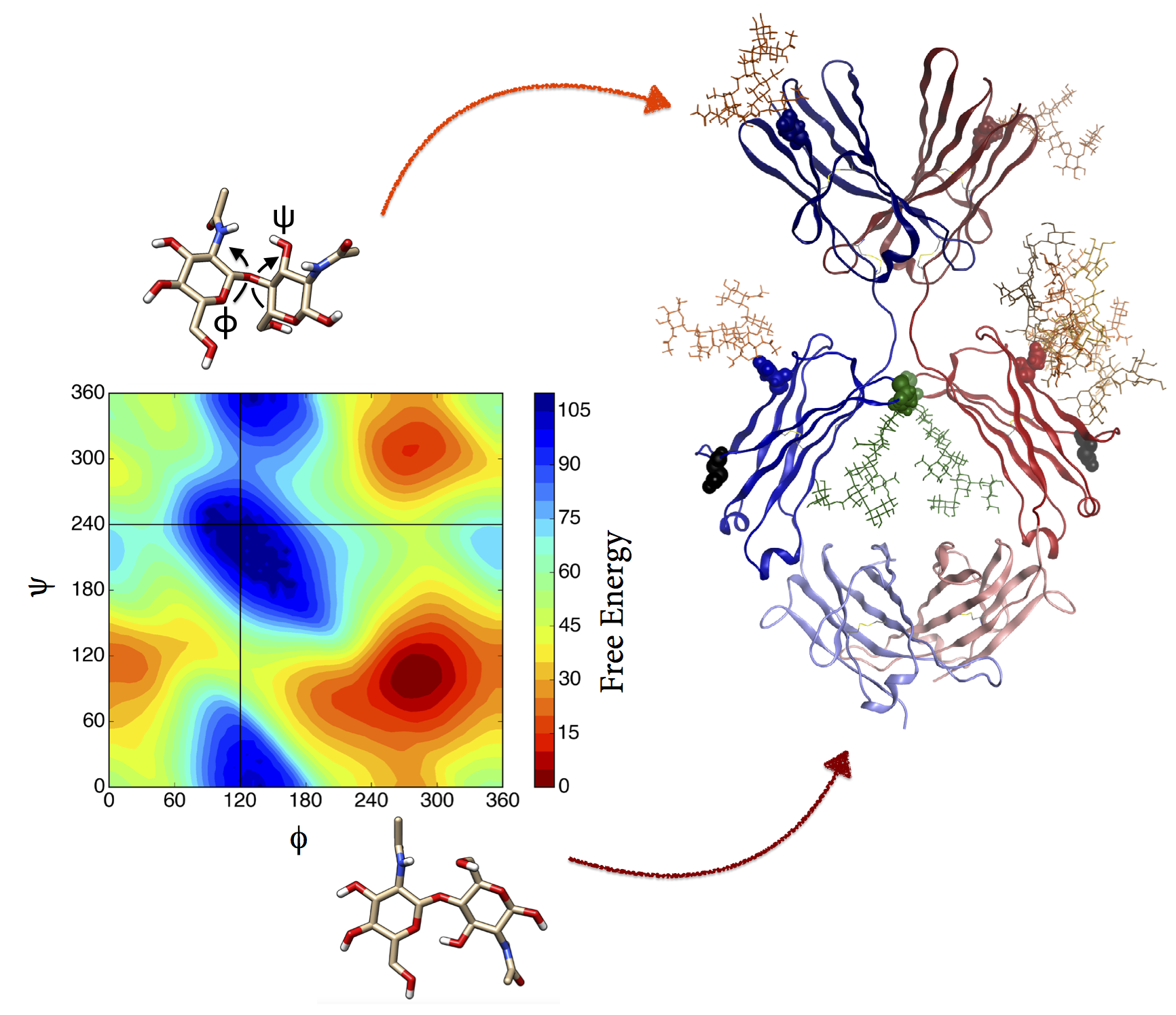
Lytic polysaccharide monooxygenases (LPMOs)
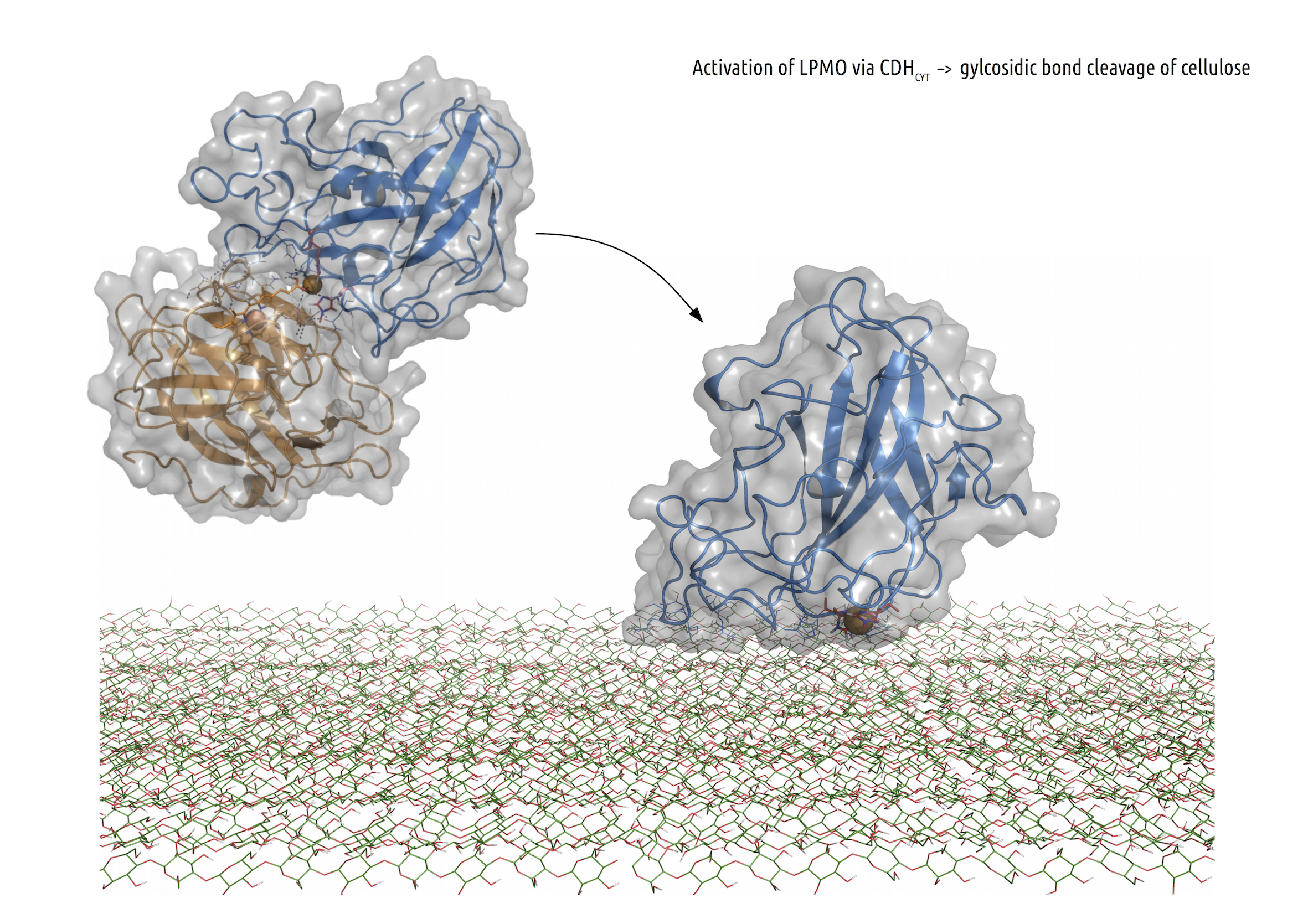
Lytic polysaccharide monooxygenases (LPMOs)
LPMOs form an important part of the cellulose, hemicellulose, starch and chitin degradation machinery. LPMOs are copper dependend enzymes, and rely on reductants like Ascorbic Acid or electron transferring enzymes like Cellobiose dehydrogenase (CDH). It is suggested that once the copper centre is reduced by one of its interacting partners, LPMOs use a copper-oxygen complex to degrade recalcitrant substrates like cellulose by cleaving glycosidic bonds. LPMO crystal structures from the fungus Neurospora crassa are used to model and simulate the protein-protein or protein-substrate interaction. Ultimately, the outcomes could help to improve the enzymes for industrial applications.