The structure and function of complex macromolecular entities, like proteins, polymers or mineral structures may be studied by molecular simulation and lead to insight into the molecular mechanisms that underlie experimental observations. Two aspects are commonly distinguished that play crucial roles to get to meaningful results:sampling and scoring. Sampling involves the generation of a sufficiently large amount of molecular structures to describe the orientations and conformations that in reality lead to the observed (average) quantities that are measured. These may be obtained from a variety of search techniques, like molecular dynamics simulations. Scoring, on the other hand, is required to evaluate how likely the generated conformations are in terms of energy and to discriminate ‘good’ conformations from ‘bad’ conformations. In general, all chemical processes are governed by the free energy differences associated with them. For this reason, free energy calculations have our particular interest. Please, browse the categories of our scientific research below to learn more about our work.
Free energy calculation

Methodology and applications of free energy calculations All chemical equilibria are governed by the difference in free energy between various states. Examples are free energy differences between different conformations of a biomolecule or the free energy difference between the bound and unbound state of a ligand and its pharmaceutical target. In our group we try to develop methods to efficiently and accurately calculate free energy differences for such processes, using statistical mechanics and molecular simulation. In recent years, we have used the one step perturbation method to calculate relative binding free energies for series of compounds. This method is currently being further developed on a set of trypsin inhibitors and applied to a crop protection target.
GROMOS

Our group contributes to the development of the Groningen Molecular Simulation (GROMOS) software package for biomolecular simulation.
Force-field development
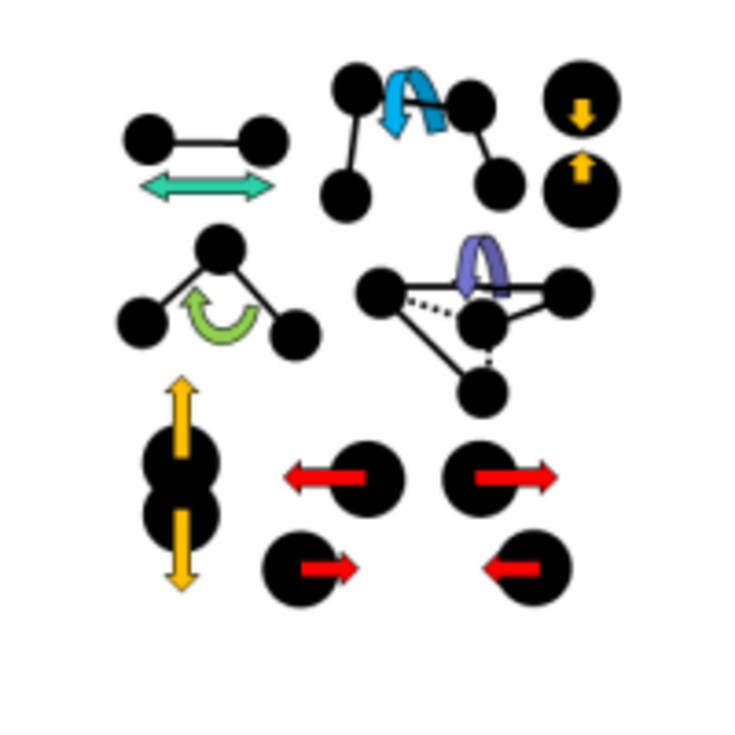
Force-field development The careful optimisation of force field parameters is essential for any classical simulation. Nonbonded interactions can currently be parameterised against free energies of solvation in various media. For ionic groups, there are several conceptual difficulties that need to be overcome, in which great progress has been made recently.
VSOMM
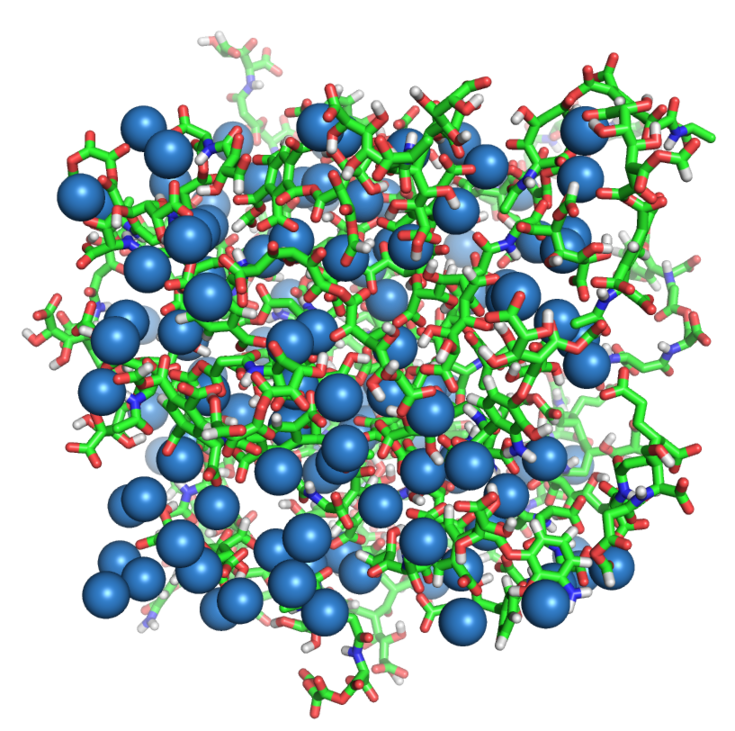
The Vienna Soil-Organic-Matter Modeller is an online tool to create condensed phase models of humic substances.
Simulation of biomolecules
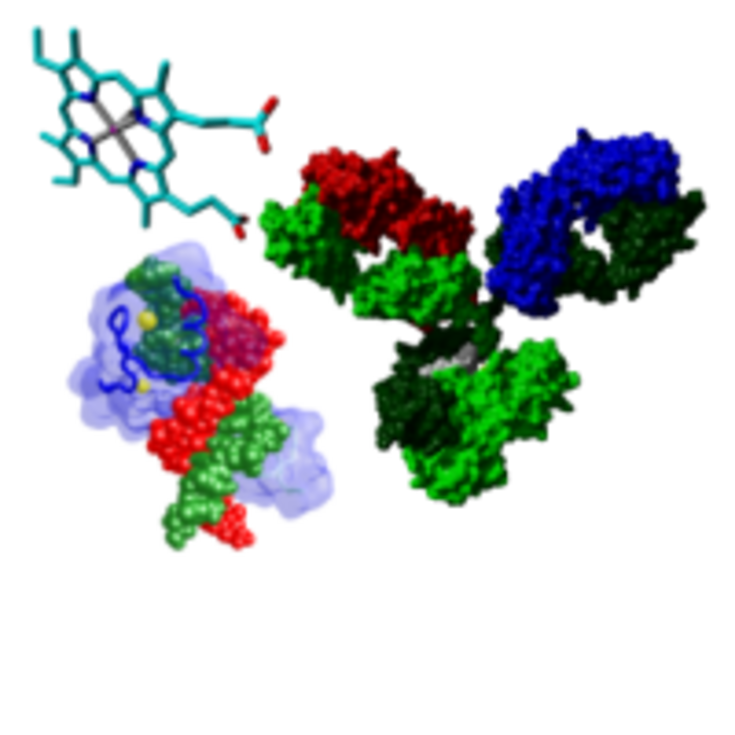
Structure and dynamics of biomolecules The structure and dynamics of complex biomolecular structures are studied by molecular dynamics simulations. This gives insight into the function, stability and potential modification sites of such molecules.
SARS-CoV-2 related research
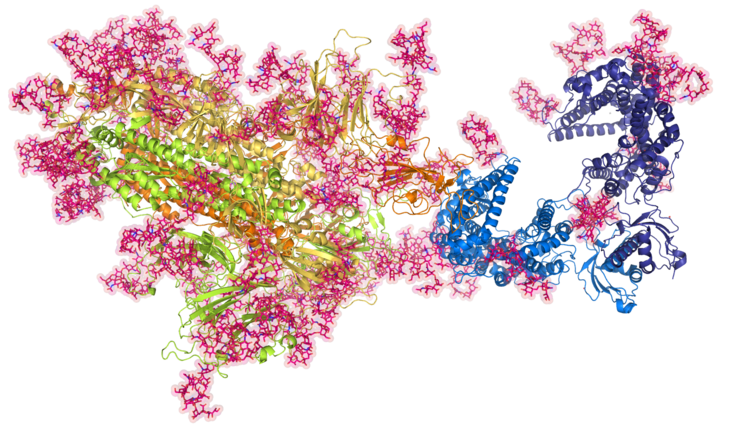
In the context of a WWTF-funded research project, we create computer models of the SARS-CoV-2 Spike protein in complex with the human ACE2 receptor. A special focus is placed on the inclusion of glycan structures.